Research
Publications
Boosting Prior Knowledge in Machine Learning for Biomarker Discovery
The so-called small n, large p problem makes learning classification or regression models difficult for biomedical applications. In such a problem especially in clinical dataset, adding samples is not easy so feature selection or embedding techniques should be used to reduce the number of features p. Prior knowledge is fundamentally important and relevant information that can provide an efficient way to regularize the number of features for training machine learning models. Because these knowledge comes from accumulated studies of various biological systems, it is also applicable to patients and model organisms.
In this study, we tried to prevent overfitting of the model and improve the robustness of the model by iteratively querying the appropriateness of the features for each model training process.
Disease Gene Prioritization for Classification (DGP4C)
Disease gene prioritization methods such as network propagation and similarity profiling have been used to prioritize candidate disease genes for experimental validation. However, it is not clear whether these disease gene prioritization methods can be used as feature selection techniques for gene expression-based disease classification models. In this study, we developed and validated a method for prioritizing disease genes that shows high importance for the classification of disease phenotypes.
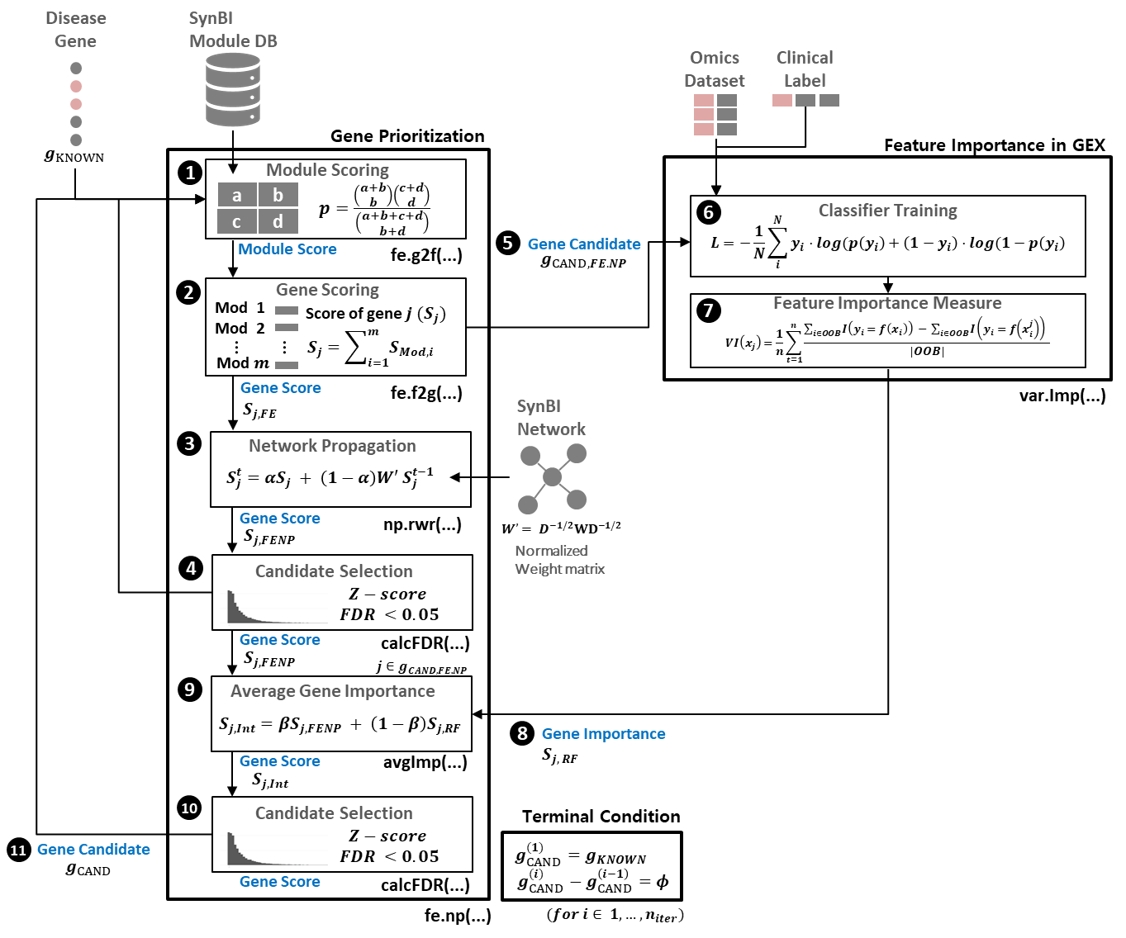
The above figure provides a schematic diagram of the proposed DGP4C framework. The algorithm begins finding candidate disease genes using a disease gene prioritization method based on a combination of disease gene scores calculated from network propagation and function similarity profiling. The feature importance of candidate disease genes for disease phenotype classification is then assessed and important candidate genes are fed back to the gene prioritization step to update candidate disease genes until no additional disease gene candidates are found.
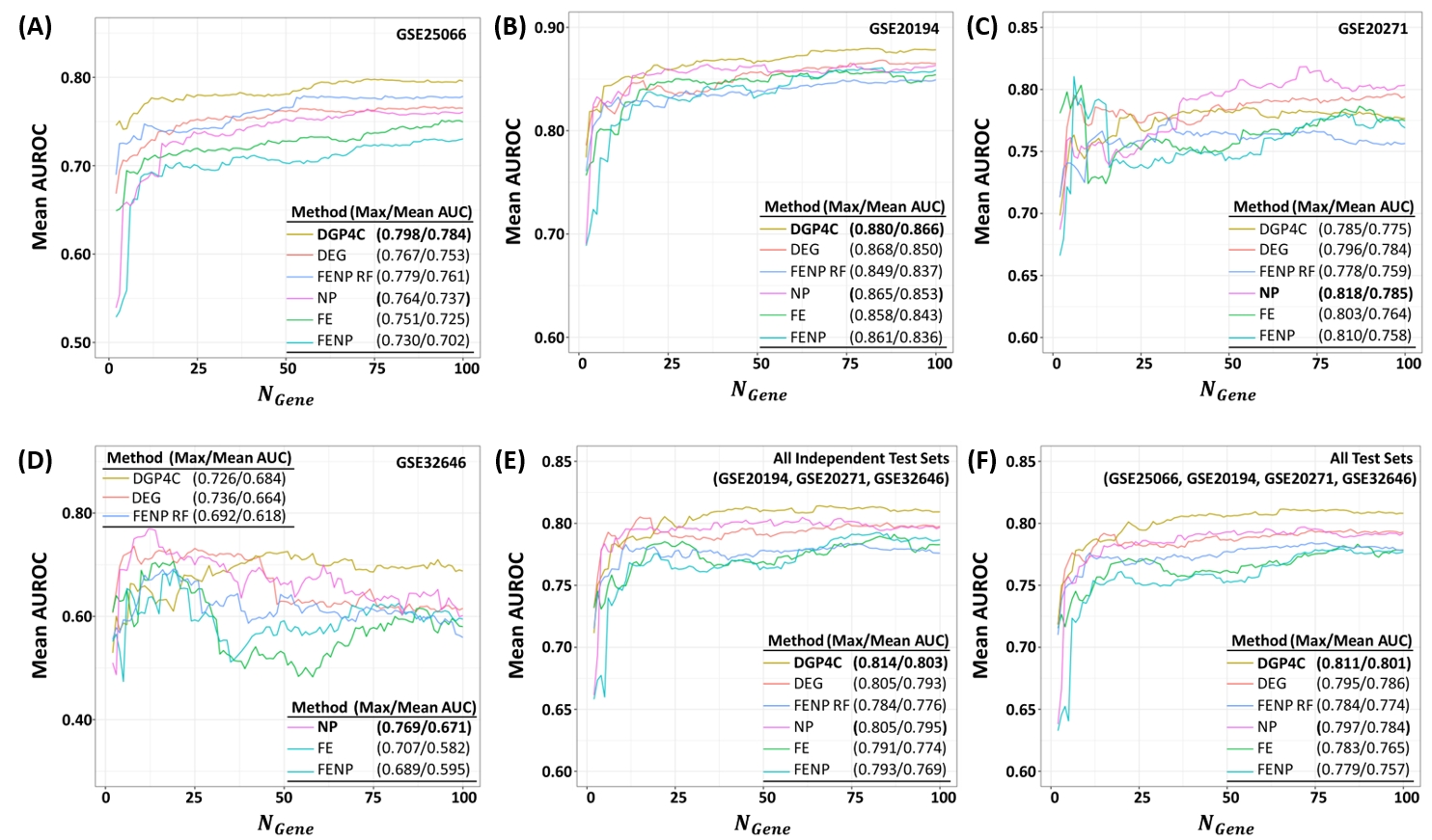
The figure shows performance evaluation result of DGP4C algorithm in Neo-Adjuvant Chemotherapy (NAC) response prediction in breast cancer. As expected, DGP4C showed best performance compared to all other non-iterative approach to optimize gene expression markerset.
Disease Mechanism Marker Optimization for Classification (DMMOC)
In many disease phenotype classification tasks, the identification of subgroups involved in disease mechanisms is an important subtask in understanding disease pathogenesis. unlike other studies that have used some clustering techniques to find subgroups, here we use iterative error estimation to find classifiable subgroups within a random feature subspace of mechanism associated feature space. For each important disease mechanism, we find associated sample sets to be used as dictionary for the new sample classification. This framework is called Disease Mechanism Marker Optimization for Classification (DMMOC) and the new test sample is classified by specific mechanism associated classifier which has higher probability of succesive classification for the test sample.
Random Subsample Subspace Ensemble (RSSE)
In the framework of the Random Subspace Ensemble (RSE) learning, a base classifier is trained on a random subspace and the informative weak classifier is determined by a selected calibration metric. The classification of new samples is performed by a majority vote in the set of informative weak classifiers. However, in many cases the RSE framework derives a set of weak classifiers that give similar decision values for the test sample. To increase heterogeneity of weak classifiers, here we proposed Random Subsample Subspace Ensemble (RSSE) learning framework. In RSSE, weak learners are trained on the subset of training samples and informative weak classifiers are selected not only based on the calibration metric but also heterogeneity of the decision values. In addition, the RSSE framework is combined with DGP4C and DMMOC algorithms to learn disease mechanism associated random subspace ensemble model. This can improve the performance of the ensemble classifier as well as the interpretability of the model.
Self-Supervised Learning for reproducible research
The self-supervised Learning (SSL) is defined as a machine learning strategy that can be supervised by the transformed training features rather than labels. Self-supervised learning is especially useful when the measurement or labeling process involves uncertainty. In biomedical dataset as well as biosignal processing, many problems can be formulated as self-supervised learning problems.
In this study, we tried to develop applicatin specific self-supervised learning models for improving reproducibility of specific applications.
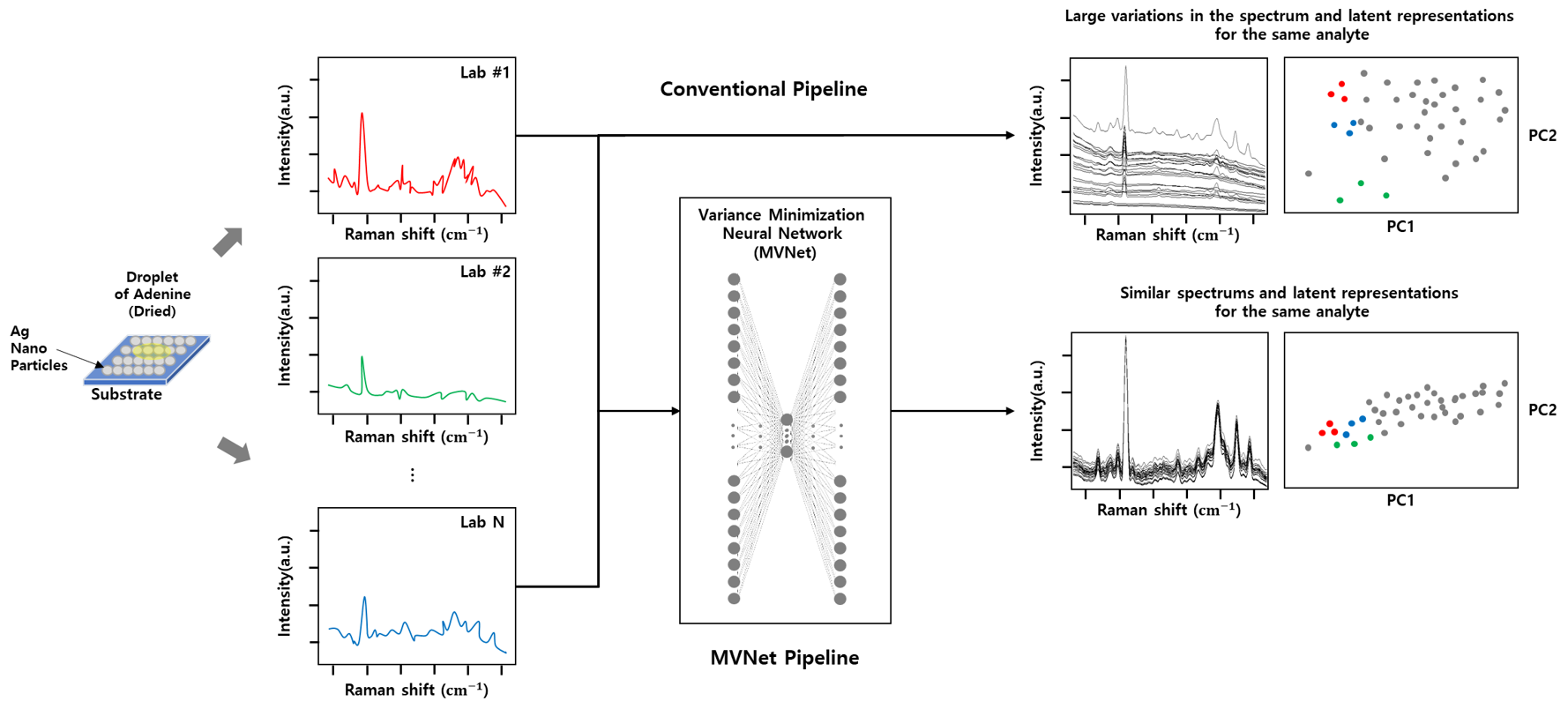
Minimum Variance Representation Learning (MVNet)
Despite the numerous advantages of surface enhanced Raman spectroscopy (SERS), it is still considered to be poorly reproducible and not a sufficiently robust analytical technique for routine implementation outside of academia. Here, we propose a self-supervised learning model for minimizing inter-laboratory variability in quantitative SERS measurements coined as MVNet. The proposed MVNet is designed to generate minimum variance SERS spectra of the same target analyte from various laboratory data while maintaining the characteristics of the original spectra. By comparing the regression performance of the generated and original SERS spectra, we showed that the proposed model can reduce interlaboratory variability while improve regression performance.
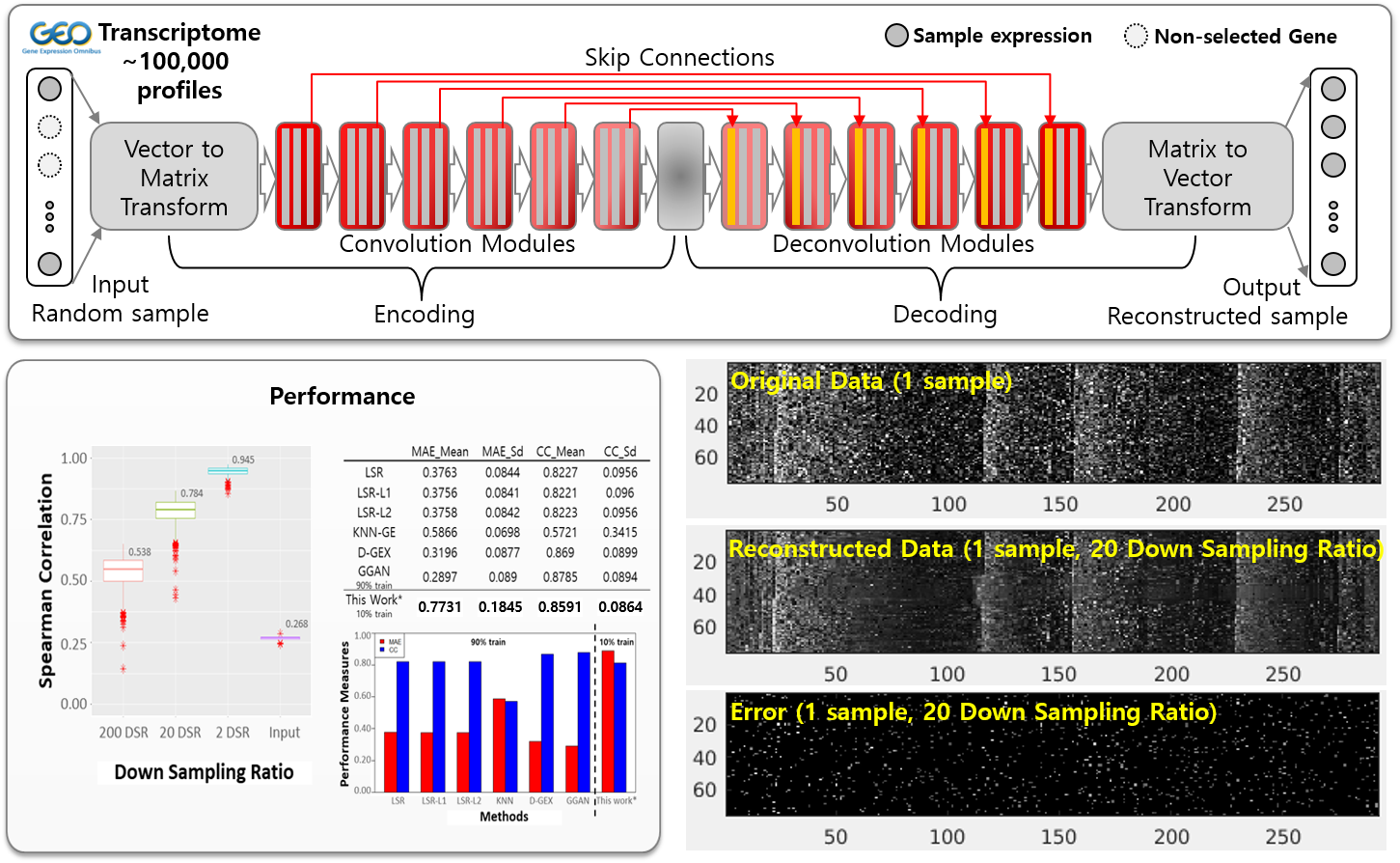
Sparse Subsample based Gene Expression Reconstruction
Cells do not express genes individually, but as a set of individual programs, and each cell expresses only a limited number of programs at a time. Due to these modular and sparse gene expression properties, it is possible to reconstruct whole gene expression vectors from subsamples of some genes. Here, we proposed a Convolutional Neural Network (CNN) model to reconstruct gene expression. The proposed model has a U-Net-like structure, includes encoding and decoding parts corresponding to transformations and inverse transformations of the data, and learns how to minimize reconstruction errors under given sparsity conditions through training. Using only 10% of the original data, this model was able to achieve 0.86 PCC values comparable to state-of-the-art gene expression reconstruction models.
Sparse Representation Learning
In many biological applications, the large number of features often makes it difficult to build a good performing classification model. Here, I and my colleagues introduced and characterized several sparsity promoting deep learning models to learn low-dimensional representations for use in downstream tasks. As an application, we applied our model for sequence based protein classification and protein-protein interaction prediction tasks.
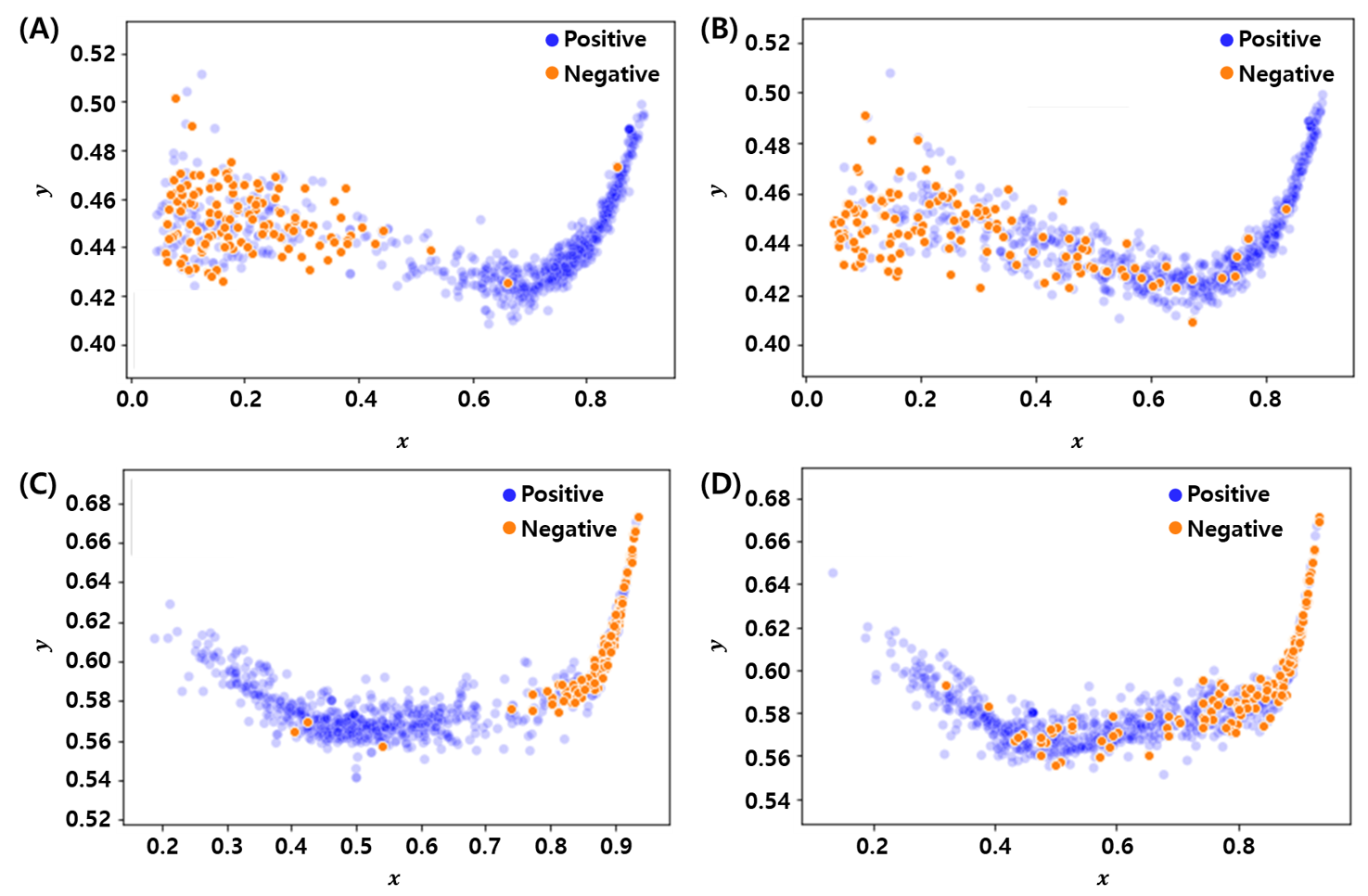
Deep Latent Space Encoding (DeepLSE)
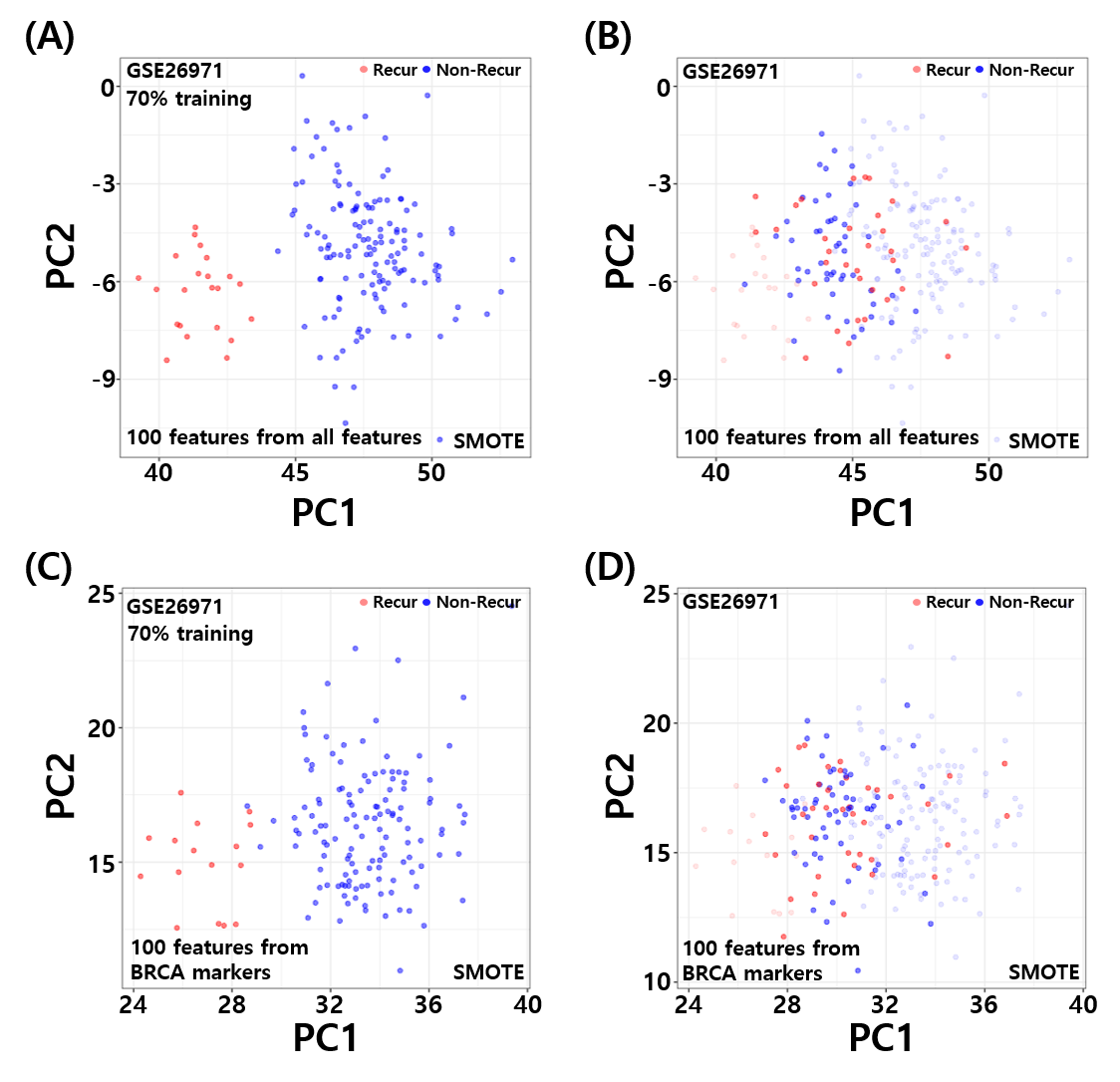
Deep Latent Space Encoding model has an ability to learn the non-linear embedding of the features whilst maintaining the properties of the original features through reconstruction. This consistency helps filter out fluctuating data points by evaluating their distance from the class centers. As a result, the model can be an important tool for designing a classifier with noisy annotations. Another advantage of using such configuration is the elimination of the need of a crucial step of feature engineering, which involves the selection of dominant features for the training of classifier. We applied DeepLSE models for several applications such as classification of Anti-Freeze Protein (AFP), Anti-Oxidant Protein (AOP), E3 and Target Pair Prediction (E3TargetPred), and Anti-Cancer Peptide (ACP) prediction tasks. The figure shows learned latent space for classification of E3-target pairs and non E3-target pairs. This visualization clearly shows that the model effectively learn low-dimensional represnetation for classification between actual E3-target pairs and Non-E3-target pairs.
Deep Sparse Reconstruction Classifier (DeepSRC)
Sparse Representation based Classification (SRC) framwork is based on a parsimonious principle in which a test sample can be reconstructed using a linear combination of over-complete dictionary matrix (ODM) elements with sparse weights. It is very robust to redundancy, because it only selects a few among all of the basis vectors. It is also very robust to noise. The DeepSRC is an autoencoder-based neural network approach that mimics the conventional SRC approach. In particular, it is composed of an encoder, a sparse coding layer, and a decoder to learn sparse representation from original features.
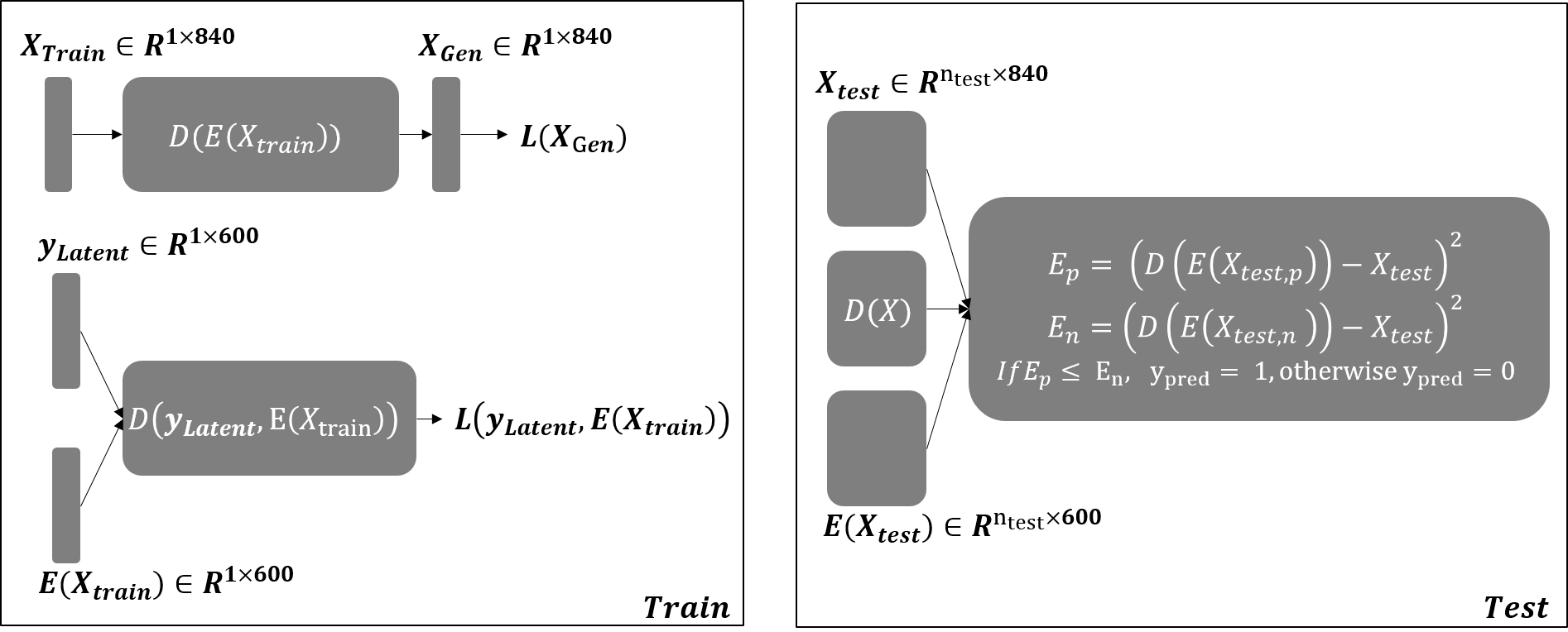
Overlap Statistics and its applications
Distribution overlap between two probability distributions are fundamental effect size measure for binary classification task. I and my collagures utilized overlap statistics as a metric for evaluating data and model in various applications, such as bioassay, differential expression/activity analysis, data/model quality assessment tasks.
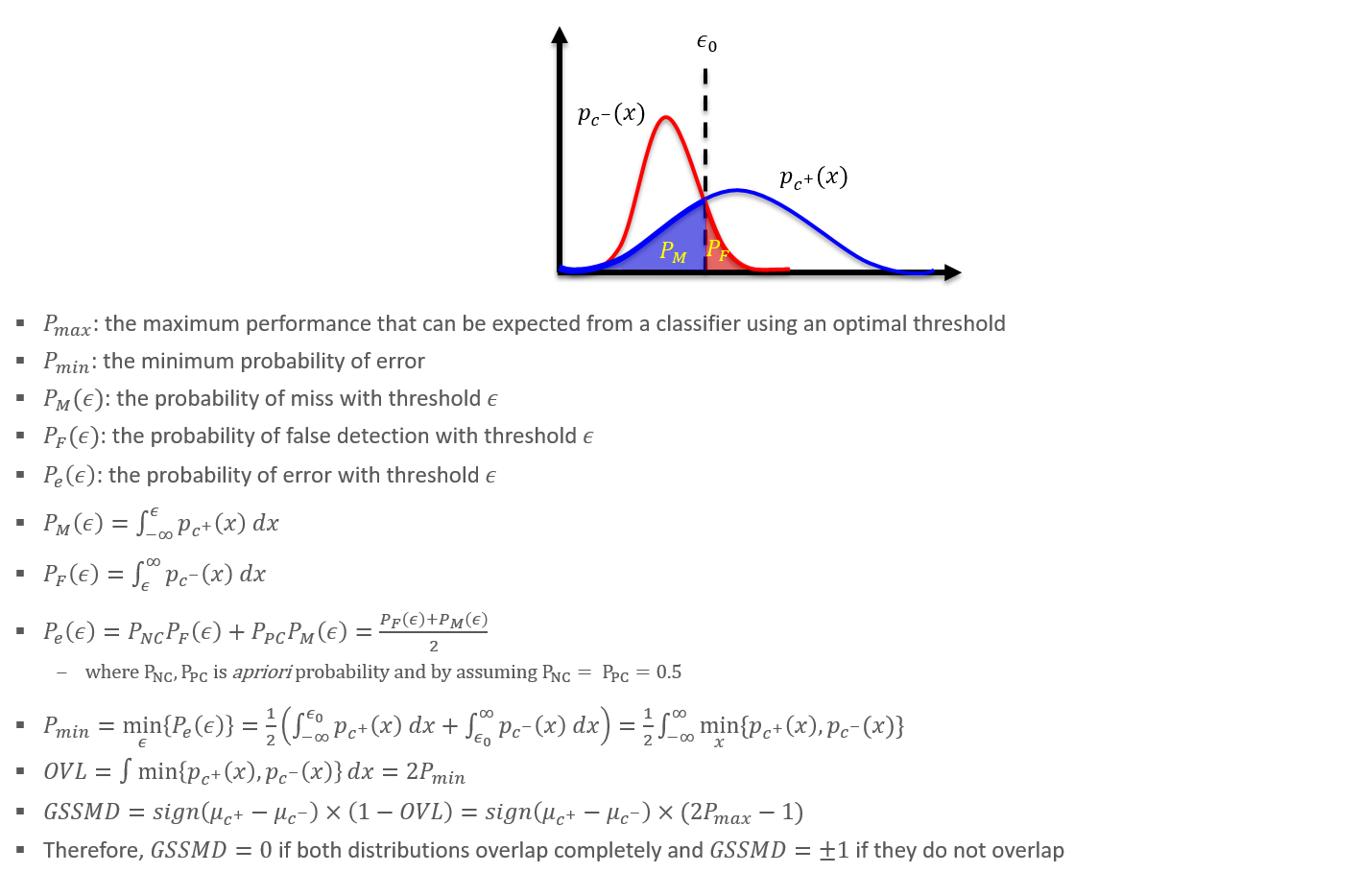
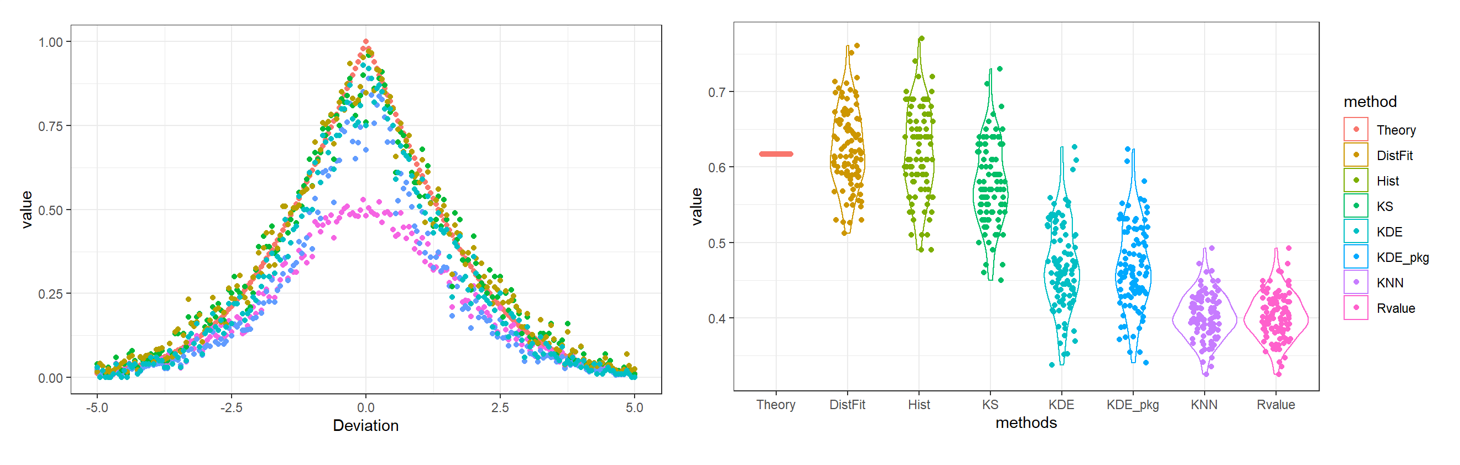
Ovltools: an R package for estimation of arbitrary distribution overlap
ovltools designed to provide some of functions that can calculate distribution overlap between two numeric vectors. It also provides significance testing based on permutation, to assess whether given sample distributions are drawn from the same distribution or not. Current version of ovltools provides KNN (K-Nearest Neighbor), KDE (Kernel Density Estimation), histogram, distribution fitting based overlap estimation methods.
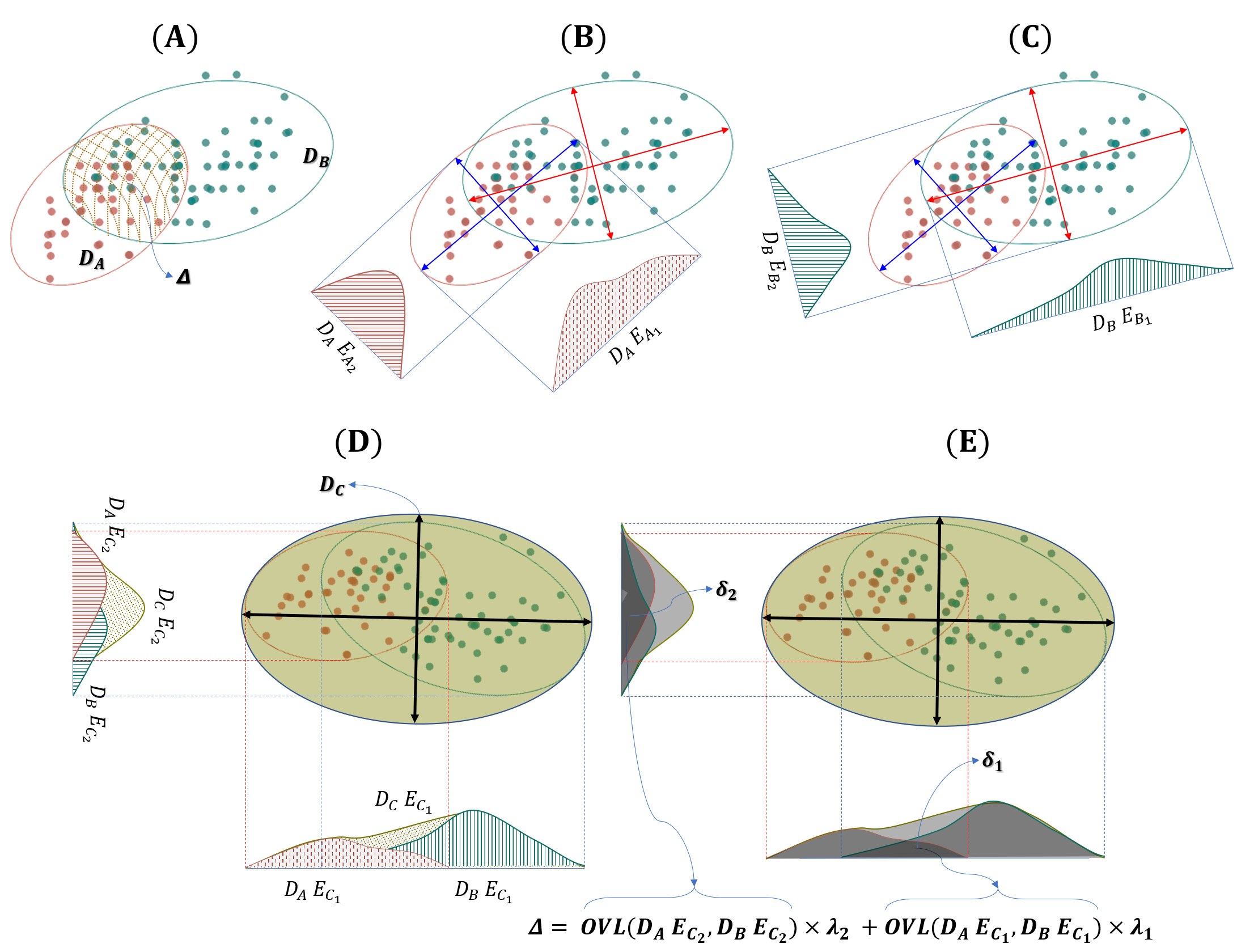
GMDM: Generalized Multi-Dimensional overlap Metric
In this paper, we extended the idea of one-dimensional overlap coefficient into a multidimensional overlap metric coined as generalized multi-dimensional distribution overlap metric (GMDM). We apply the GMDM to evaluate the quality of the style-transfer model trained in an unsupervised fashion, for a denoising model trained in a supervised fashion, and the class separability of different image datasets. We also apply the GMDM to assess the classifiability of different protein sequence encoding schemes for the antioxidant protein classification problem.