
Research
My research develops computational methods that read biological signals from high-dimensional measurements — gene expression, spatial transcriptomics, Raman spectra, phonocardiograms — and grounds them in explicit biological priors or physically meaningful summary statistics. Four connected threads run through the work:
- Cell–cell communication and cytokine-activity inference
- Clinical biomarker discovery with biological priors
- Sparse representation and overlap statistics
- Biosignal processing and reproducibility
See the Publications page for a full list; Google Scholar is kept current.
Cell–cell communication and cytokine-activity inference
A through-line from wet-lab beginnings. My earliest work (2011–12, with Taesung Kim’s group) built microfabricated concentrator arrays and inkjet-printed bacterial cell systems to study synthetic bacterial cell-to-cell communication and predator–prey dynamics at single-cell resolution (Biomaterials 2011; Lab on a Chip 2011, 2012). The underlying question — how do cells exchange signals, and can we read those signals quantitatively? — is the same question I now address computationally in human tissue.
At NCI/CDSL, I contribute to and develop tools that infer the activity of secreted proteins and cytokines from transcriptomic data:
- secactpy — Python package for secreted-protein activity inference from bulk and single-cell gene expression; hosted under the Jiang Lab
data2intelligenceorganization. (Author.) - spatial-gpu — GPU-accelerated spatial kernels for neighborhood activity estimation on spatial transcriptomics. (Author.)
- ridger — R package for ridge-regression-based activity inference. (Main contributor; project initiated by B. Ru.)
Related R packages from the Jiang Lab that I’ve contributed to: secact and spacet (Spatial Cellular Estimator for Tumors).
Additional resources and manuscripts in this area are ongoing at NCI/CDSL and will be linked here upon release.
Clinical biomarker discovery with biological priors
Classification and regression for biomedical phenotypes suffer from the small-n, large-p regime: patient cohorts are small, features (genes, peptides) are many, and labels are expensive. Prior knowledge of disease mechanisms — pathway memberships, interaction networks, causal hypotheses — provides an efficient way to regularize and interpret these models.
IC2Bert — immune-checkpoint-blockade response prediction
Masked-gene-expression pretraining followed by supervised fine-tuning for robust prediction of response to immune-checkpoint-blockade therapy (Park, Kim, Jiang, Scientific Reports 2025). The model learns tumor transcriptomic representations that generalize across cohorts and treatment arms.
AI in precision oncology (co-author)
“Hallmarks of artificial intelligence contributions to precision oncology” (Chang, Park, Schäffer, Jiang, Ruppin, Nature Cancer 2025) — a conceptual review mapping where AI has genuinely advanced oncology practice versus where claims outrun evidence.
DGP4C — Disease Gene Prioritization for Classification
Disease-gene prioritization methods (network propagation, functional similarity) are usually evaluated by their ability to recover known disease genes. DGP4C asks a different question: are prioritized genes useful as features for disease-phenotype classification? An iterative loop between prioritization and model-based feature importance selects a compact, mechanism-anchored marker set.
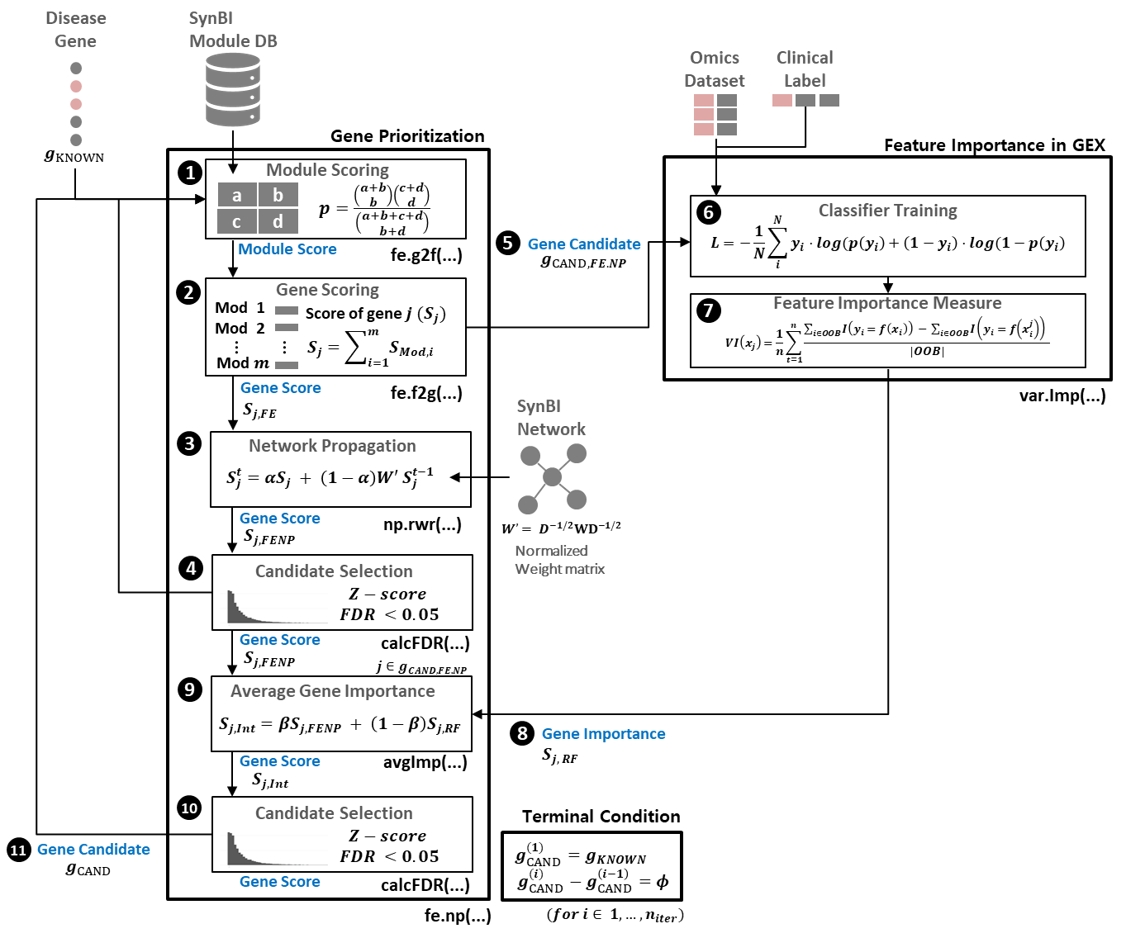
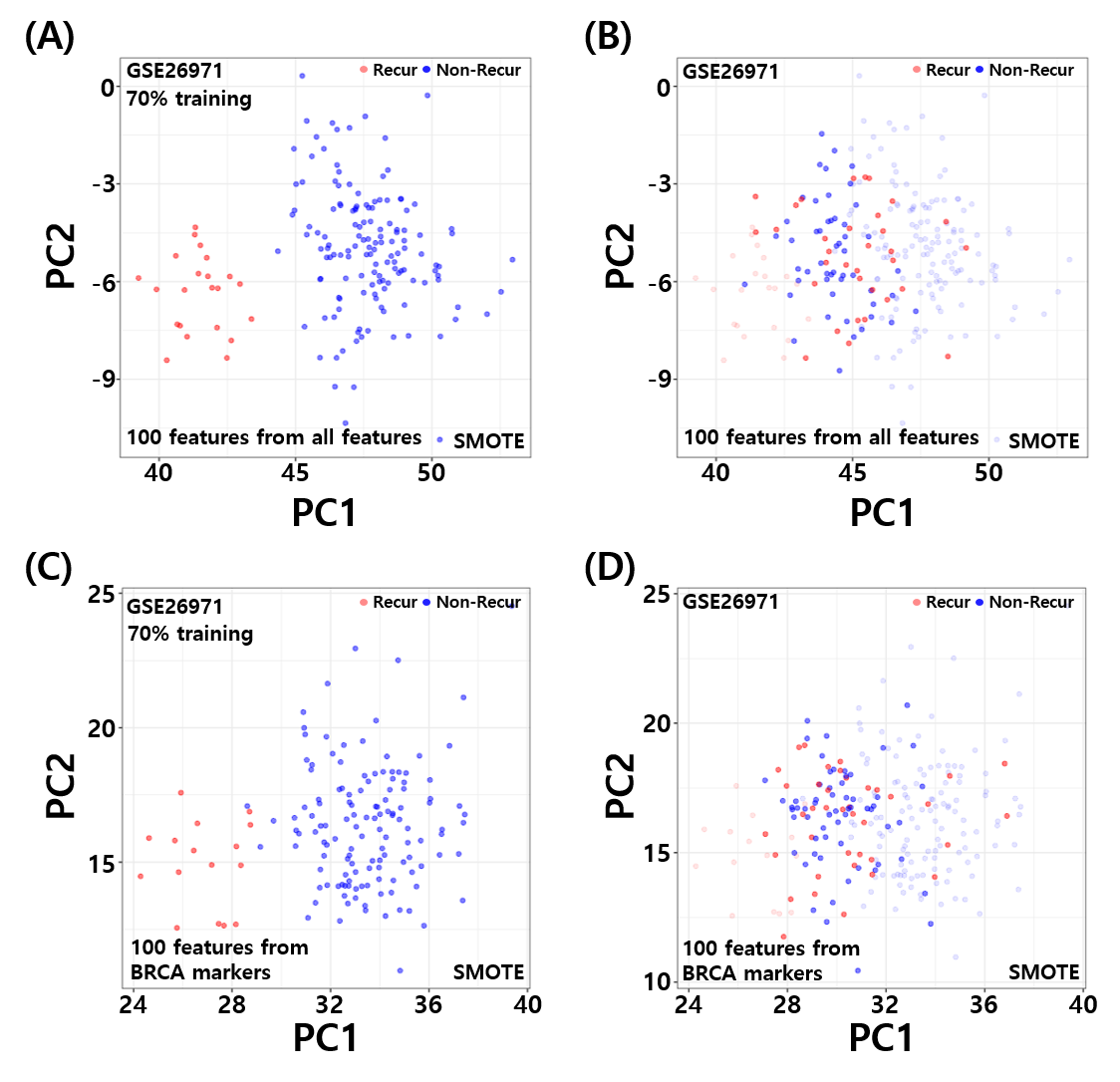
Benchmarked on neoadjuvant-chemotherapy response in triple-negative breast cancer (Park, Yi, Cancers 2022), DGP4C outperformed non-iterative feature-selection baselines.
DMMOC and Random Subsample Subspace Ensembles
Extensions of DGP4C that (i) discover classifiable subgroups within mechanism-associated feature subspaces (DMMOC) and (ii) build ensembles of weak classifiers whose heterogeneity is enforced both in feature subspace and in sample subset (RSSE). These improve interpretability and calibration of mechanism-aware disease classifiers.
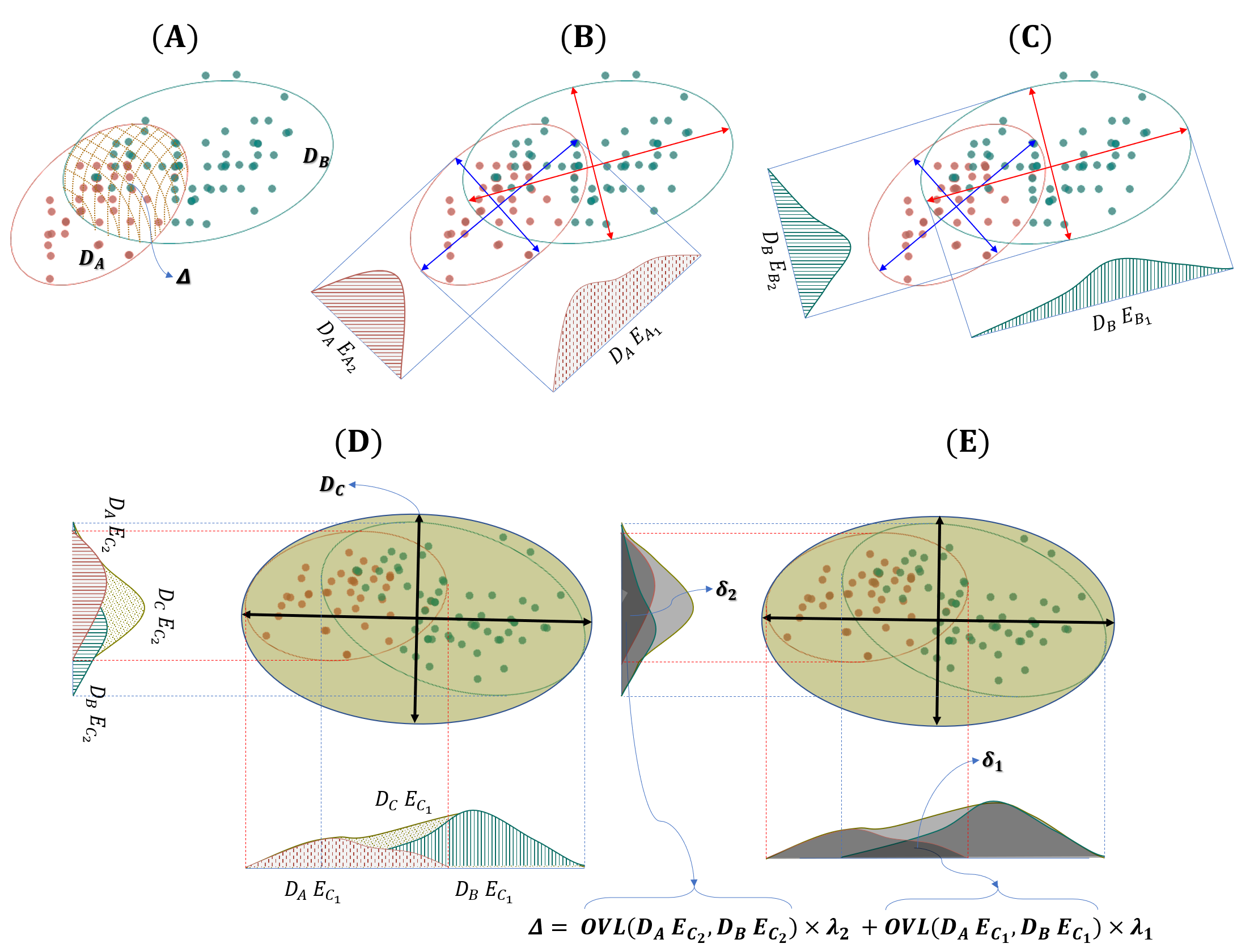
Sparse representation and overlap statistics
Two methodological threads that appear across biomedical applications.
Deep Latent Space Encoding (DeepLSE) and Deep Sparse Reconstruction Classifier (DeepSRC)
DeepLSE learns non-linear low-dimensional embeddings while enforcing reconstruction consistency, filtering noisy annotations by distance to class centers. Applied to antifreeze proteins (AFP-SRC, Neural Comput. Appl. 2022), antioxidant proteins (AoP-LSE, Curr. Issues Mol. Biol. 2021), E3–target pair prediction (E3TargetPred, arXiv 2020), and anticancer-peptide classification (IEEE Access 2023).
DeepSRC is an autoencoder realization of the sparse-representation-based classification (SRC) framework — a sparse coding layer between encoder and decoder — that inherits SRC’s robustness to redundant and noisy features.
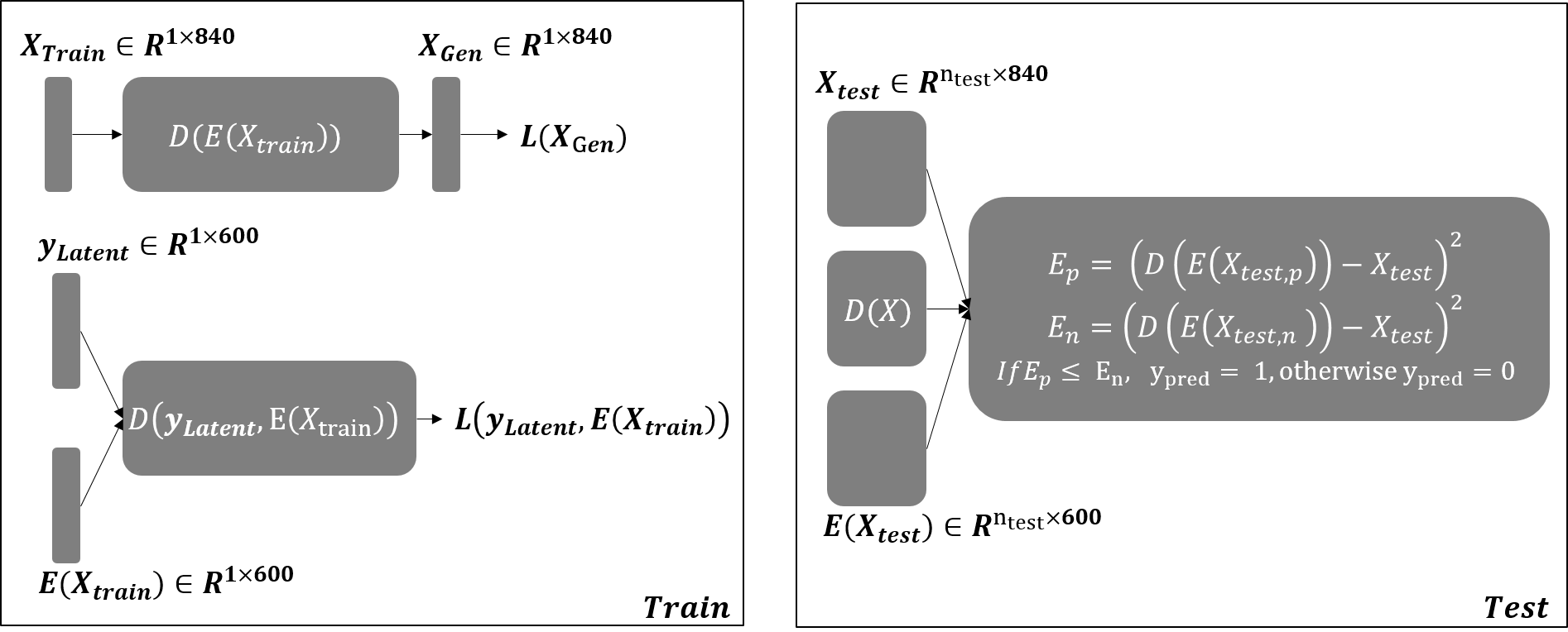
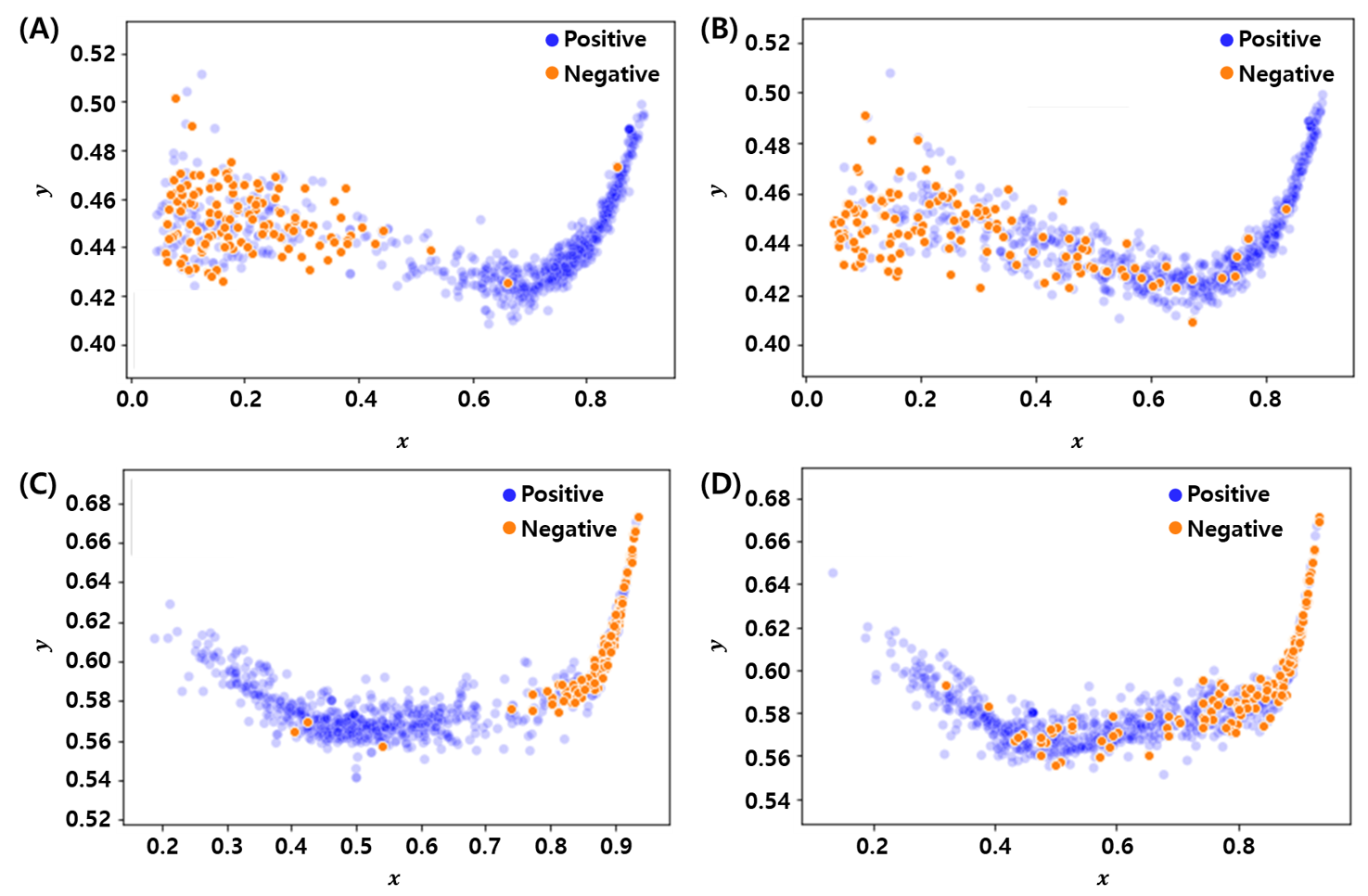
Overlap statistics: GSSMD, ovltools, GMDM
Distribution overlap is a fundamental effect-size measure for binary classification, and a principled alternative to Z-factor or SSMD in bioassay quality assessment.
- GSSMD — Generalized Standardized Sample Mean Difference, a robust effect-size measure for biological assays (IEEE BIBM 2020; arXiv 2020).
- ovltools — R package for KDE-, KNN-, histogram-, and distribution-fitting-based overlap estimation with permutation-based significance tests.
- GMDM — Generalized Multi-Dimensional overlap Metric (Digital Signal Processing 2023), extending one-dimensional overlap to multivariate data. Applied to style-transfer quality, denoising-model evaluation, and sequence-encoding classifiability.
Biosignal processing and reproducibility
Biomedical signals carry instrument- and site-specific confounds that undermine cross-laboratory reproducibility. Self-supervised learning and domain-specific architectures provide a way to separate the biology from the instrument.
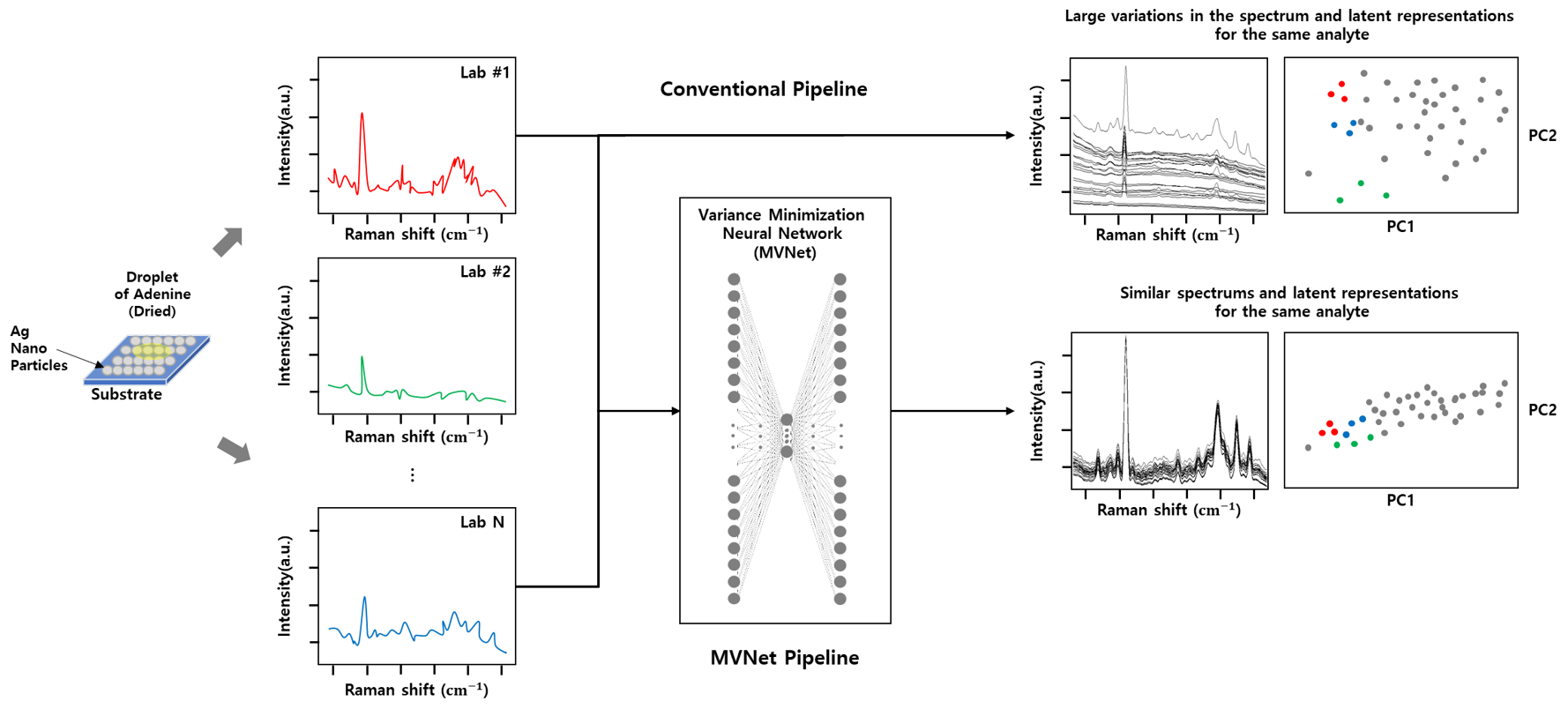
MVNet — inter-laboratory variation minimization for SERS
Surface-enhanced Raman spectroscopy (SERS) offers exquisite sensitivity but notoriously poor cross-lab reproducibility. MVNet is a self-supervised model trained to generate minimum-variance SERS spectra of the same analyte across laboratories while preserving analyte-specific spectral features (Analyst 2023).
SERSNet and ML-based SERS detection
Deep-neural-network architectures for biomolecule detection (Biosensors 2021) and heavy-metal-ion detection (Sensors 2022) from SERS spectra.
Phonocardiogram (PCG) analysis
Temporal-convolutional feature extraction with asynchronous channel-information fusion for heart-abnormality detection in multi-channel phonocardiograms (Shin et al., Comput. Methods Programs Biomed. 2025). Continuing work on foundation-model-based evaluation for PCG is ongoing.
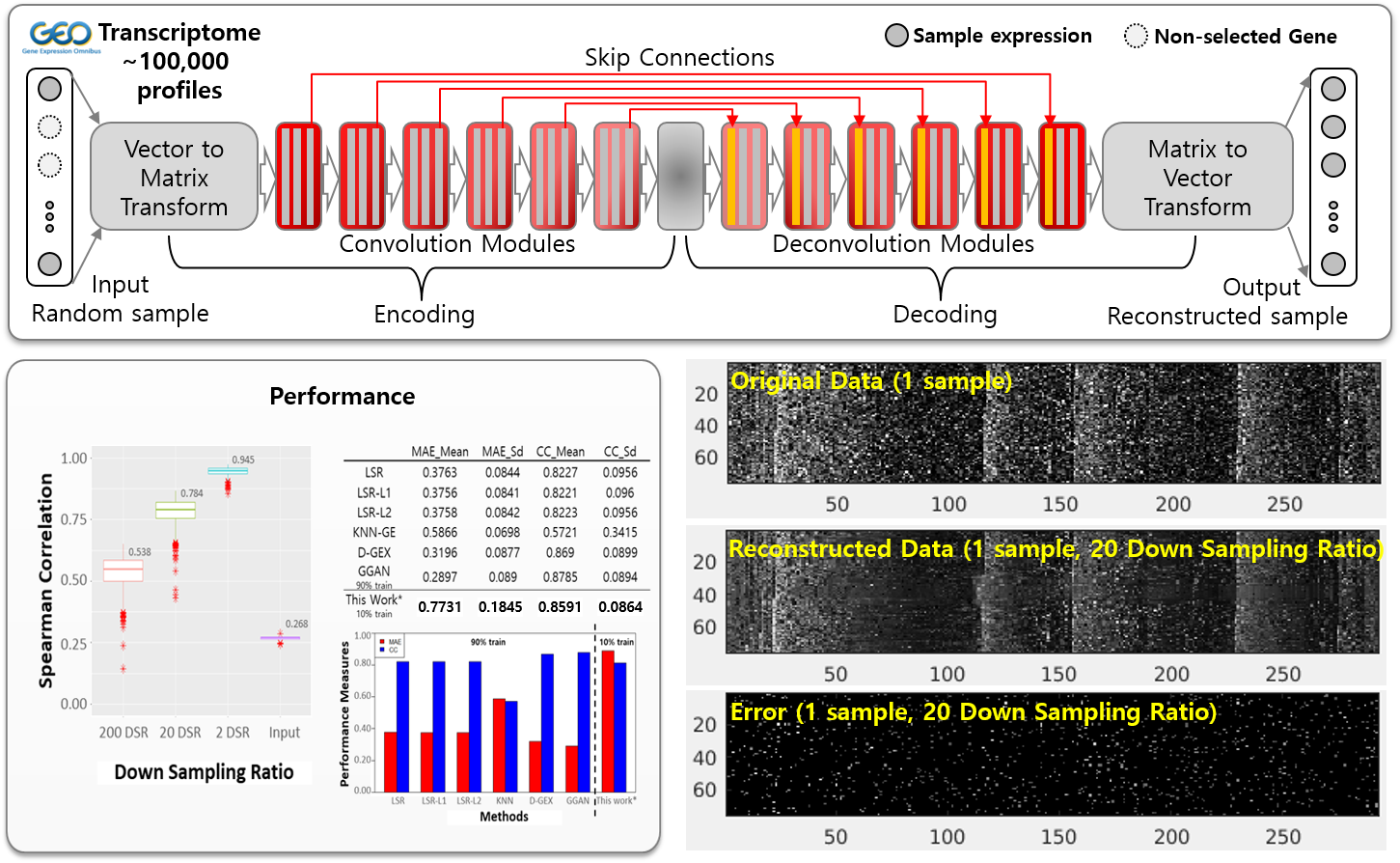
Sparse-subsample gene-expression reconstruction
A U-Net-like CNN reconstructs full gene-expression vectors from 10 % subsamples, exploiting the modular and sparse structure of transcriptional programs (PCC ≈ 0.86 versus state-of-the-art reconstruction methods).
Earlier work: microfluidics and synthetic biology
Experimental systems for studying bacterial communication and predation at single-cell resolution — microfluidic concentrator arrays (Lab on a Chip 2011, 2012) and inkjet-printed multicellular systems (Biomaterials 2011; International Journal of Molecular Sciences 2011). These are the experimental roots of my current computational interest in cell-to-cell communication.